Dr A. Gironés Muriel. H. Sanitas La Moraleja.
Dr. Angel Villar-Pellit. H. Virgen de la victoria.
Introducción
En el capitulo anterior se intentó explicar como la acción principal de estas sustancias es la de modificar la permeabilidad de las membranas nerviosas a los iones de K+ y de Na+. Siendo más sensible la afectación iónica del Na+ través del bloqueo de su canal específico. Esta acción fundamental se verá influenciada por varios factores que serán en última medida los responsables de su mayor o menor resultado clínico.
– Por un lado la cantidad de anestésico disponible propiamente dicho en su lugar de acción será el factor predeterminante para que exista bloqueo de la conducción. Sabiendo de antemano que es necesario el bloqueo de una determinada cantidad de canales para que se impida la conducción nerviosa, por tanto hay que valorar la cantidad de anestésico disponible en el lugar de acción, que no es sinónimo de la cantidad de fármaco administrado, sino la cantidad de moléculas que son capaces de llegar al axoplasma del axón.
– Por otro lado valoraremos las propias características farmacológicas del anestésico. Los mecanismos de metabolización y los propios metabolítos, la absorción, y factores como su pKa, liposolubilidad, afinidad por las proteínas, etc. Serán factores que determinaran la cantidad de anestésico activo disponible en el axón, que será siempre una cantidad menor que la administrada desde el principio.
– Y por último, el tipo y tamaño de estructuras nerviosas diana, pues como hemos visto hay fibras más o menos aisladas por la mielina, que junto a la mayor o menor profundidad de los axones centrales determinarán también la cantidad de anestésico disponible. Dicho de otro modo no es lo mismo bloquear las gruesas raíces nerviosas a nivel lumbar que a nivel torácico por ejemplo en una epidural.
Estructura de los anestésicos locales
Los anestésicos locales usados actualmente se engloban en un esquema estructural que permite estudiar sus elementos principales; la cadena intermedia, el polo hidrófilo y el polo lipófilo.
Tomado de Cousins
– La cadena intermedia o hidrocarbonada, de una longitud determinada parece ser responsable de la liposolubilidad del producto en cuanto un alargamiento de dicha cadena parece producir fármacos más liposolubles, más potentes pero también más tóxicos.
– El polo lipófilo o núcleo aromático. La estructura aromática con un anillo benzoico o paraaminobenzoico (estos últimos de importancia en los alérgicos al grupo paraaminobenceno)es en gran medida el responsable de la difusión y la fijación del anestésico.
– El polo hidrófilo, que modulará la hidrosolubilidad y por tanto su difusión sanguínea y la ionización de la molécula. De reseñar que la supresión de este polo no incide en la actividad anestésica pero limita el empleo del fármaco a aplicaciones tópicas.
– La naturaleza del enlace que une la cadena intermedia al polo lipófilo origina las dos grandes familias de anestésicos locales de la que disponemos hoy en día, las aminoaminas y los aminoesteres. La principal diferencia entre estos dos tipos de fármacos radica en su metabolización en cuanto los esteres son hidrolizados mediante enzimas plasmáticas y las amidas mediante degradación hepática lo cual convierte a estas últimas en sustancias más estables en condiciones fisioquímicas más difíciles, pudiendo por ello mezclarse con ácidos y bases fuertes y soportar mejor los cambios de luz y temperatura.
| AMINOESTERES | AMINOAMIDAS |
| Cocaína | Lidocaína |
| Benzocaína | Mepivacaína |
| Procaína | Prilocaína |
| Tretracaína | Bupivacaína |
| 2.cloroprocaína | Etidocaína |
| Procainamída | |
| Ropivacaína | |
| Articaína | |
| Levobupivacaína |
Dentro de los anestésicos amino-amidas disponemos de varias subfamilias dependiendo del ácido del cual se derivan:
– Derivados del ac. acético: Lidocaína.
– Derivados del ac propiónico: Prilocaína.
– Derivados del ac pipecólico: La mepivacaína , la ropivacaína , la bupivacaína racémica y su forma L+ o levobupivacaína
Mención aparte es la llamada articaína: Un anestésico de uso extendido en ciertos hospitales principalmente para uso en cirugía dentaria y dermatológica. Dicho fármaco es en sí una aminoamida pero tiene en su composición un grupo ester adicional que le hace tener una metabolización a través de colinesterasas típicas de esta familia convirtiéndose en un metabolito inactivo y por tanto en un anestésico de corta duración.
Nuevos Enantiómeros
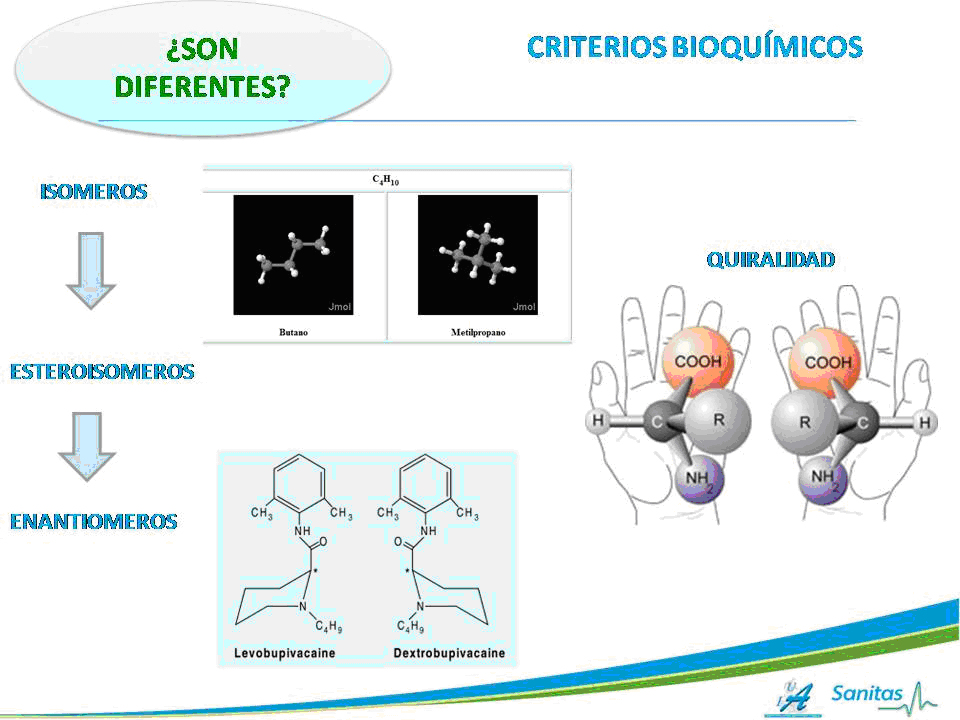
Todos los anestésicos tipo amidas de la familia pipecol (mepivacaína, bupivacaína, ropivacaína) junto a la Prilocaína y la etidocaína comparten una característica bioquímica; son moléculas quirales pues tienen en su estructura molecular un carbono que funciona como eje quiral. Para explicar este concepto explicaremos las definiciones de las moléculas en base a sus semejanzas estructurales.
Cuando dos moléculas comparten el mismo número de átomos pero no sus relaciones entre sí, decimos que son isómeros, (por ejemplo el butano y el metilpropano). Los isómeros son por tanto moléculas con propiedades físicas y químicas que pueden ser muy distintas entre si.
Cuando la relación de los átomos intrínsecos de las dos moléculas es también la misma, hablamos de esteroisómeros, que sí tienen parecidas propiedades físicas y químicas. Ahora bien, si esos esteroisómeros tienen un eje desde el cual la molécula puede rotar adoptando dos formas como si estuviesen enfrentadas en un espejo hablamos de enantiómeros o moléculas quirales, diferenciándose por tanto dos formas moleculares; la forma levógira o L y la forma dextrógira o S.

La quiralidad es un concepto más espacial que químico, su definición viene del griego, (“Quiros“ que en griego es manos) y como las manos, las estructuras quirales son objetos simétricos, no superponibles, y por tanto no iguales. Esto significa que de todos los anestésicos con capacidad quiral (mepivacaína y bupivacaína principalmente) la mayoría se nos ofrecen en una mezcla de dos tipos de fármacos: la forma levógira y la forma dextrógira de la molécula al 50% conjuntamente en una presentación llamada mezcla racémica.
La importancia de presentar un anestésico mezcla racémica o depurada es muy importante. De hecho la depuración de las moléculas quirales en nuestro medio se ha hecho desde el principio por la propia naturaleza.
Toda la arquitectura celular que existe en nuestro querido planeta conformada por los 20 distintos aminoácidos (que son también enantiómeros) son levógiros o formas L. Tan solo en un laboratorio se pueden encontrar aminoácidos con forma D. El adn y la glucosa son en cambio dextrógiras. Esto condiciona una premisa muy interesante; que aunque los enantiómeros presenten las mismas características fisico-químicas, sus propiedades farmacodinámicas y farmacocinéticas son diferentes puesto que las estructuras enzimáticas están preparadas principalmente para formas L.
Esto condiciona un concepto nuevo;“ suciedad racémica“ en la que básicamente nos indica que al usar este tipo de fármacos estamos dando un 50% de producto útil, condicionado por que la forma dextrógira restante del anestésico es una forma más tóxica y no aporta beneficios al paciente.
Desgraciadamente no es raro encontrar en nuestro medio cierta resistencia al uso de los nuevos enantiómeros por parte de algunos hospitales. La supuesta falta de estudios claros en humanos y en fetos no es una razón de peso que enmascara razones fármaco-económicas que no deben de apartarnos de nuestra labor fundamental como médicos, que es la de proporcionar los mejores cuidados disponibles al paciente. El estudio razonado de los ensayos en animales, la comprensión de su mecanismo de acción desde un punto de vista exclusivamente farmacológico y la visualización del número de casos registrados de toxicidad en humanos, cuyo interés empezó tras una publicación en 1975 de la editorial Albright. Así como las diferencias constatadas en la evolución clínica de los casos de sobredosis según el anestésico utilizado nos avala en nuestra postura de usar estos nuevos anestésicos.
De todo ello hablaremos más adelante con los anestésicos“ depurados“ que la industria farmacéutica nos brinda actualmente: el Chirocane© como la forma L depurada de la bupivacaína, y Naropin© como forma depurada L de la ropivacaína.
Metabolismo de los anestésicos
El modo y velocidad de degradación tiene una importancia no solo para valorar los márgenes de seguridad del fármaco, si no que marcan el tipo de anestésico usado y muchas de sus implicaciones clínicas tienen que ver con su metabolización.
Los anestésicos tipo ester sufre una degradación enzimática a nivel sanguíneo mediante las colinesterasas plasmáticas, esta condición a priori origina una rápida metabolización de los mismos, sin embargo esta misma característica es en parte la responsable de su abandono por parte de anestesiólogos actuales.
Por una parte los enfermos con un déficit enzimático adquirido (en hepatopatías) o un déficit congénito de colinesterasas podrían desencadenarse toxicidades tanto a nivel local como general, del mismo modo también existe la posibilidad de generarse bloqueos neuromusculares prolongados por agotamiento de la colinesterasa , situación ésta que podríamos encontrarnos en administraciones de uso continuado en el tiempo como se puede dar en tratamientos de dolores crónicos o cualquier actuación que requiera una infusión de anestésico de manera continua y prolongada.
Otra característica del metabolismo de los esteres que originó el abandono por parte de los anestesiólogos actuales son los cuadros alérgicos a sus metabolitos. Estos aparecen bien por alergia directa (principalmente al grupo paraaminobenceno ) o bien reacciones alérgicas cruzadas (como en los alérgicos a las sulfamidas) .Tendremos en cuenta no obstante que ciertas amidas tienen un estabilizador o conservante , el metil o butil-paraben cuya estructura se asemeja a la del grupo paraminobenzoico que explicaría ciertas reacciones cruzadas entre aminas y esteres, que aunque raras existen.
Los anestésicos tipo aminas en cambio tienen una metabolización hepática. El famoso citocromo P450 es el principal implicado en dicha metabolización. Dentro de esta vía de metabolización nos encontramos con la isoencima CYP 1A2 que representa tan solo el 10% del citocromo P 450, pero que en cambio tiene una gran afinidad por determinados anestésicos, sobre todo para la mepivacaína y la ropivacaina . La existencia de esta isoencima CYP1A2 es dependiente de la edad del sujeto lo que obliga a replantearse el uso de la mepivacaína y la ropivacaina en recién nacidos, puesto que este tipo de pacientes presentan una tasa de esta isoencima baja, lo cual origina una vida media de eliminación mayor respecto a la del adulto y por tanto mayores posibilidades de alcanzar dosis tóxicas.
En cambio la isoencima CYP 3A4 se presenta en grandes cantidades en el ser humano y aunque su afinidad sea menor por los anestésicos, existe una tasa en el recién nacido similar a la del adulto por lo que son vías difícilmente saturables y poco dadas a la interacción metabólica. Anestésicos con preferencia por esta vía son la lidocaína y la bupivacaína.
A la luz de estos hechos nos preguntamos cual es el perfil de seguridad exigible a los anestésicos usados en obstetricia y en pediatría y deberíamos valorar si las diferentes tasas de metabolización son suficientes para exigirnos cambiar de anestésico según el paciente.
A nivel puntual cada aminoamida presenta ciertas características en su metabolización que también pueden ser interesantes, en cuanto podrían modificar su uso clínico dependiendo del paciente y el tiempo de administración. Apuntamos algunas.
LIDOCAíNA
Se metaboliza en el hígado principalmente, biotransformándose en un metabolito biológicamente activo (el MEGX y su derivado; la xilidide de glicina) con actividad cardiovascular y epileptógena. Es decir, hay que valorar que sus efectos a nivel cardiaco y de sistema nervioso central pueden acumularse en administraciones repetidas y continuas puesto que estos metabolitos resultantes como la xilidide de glicina tiene una excreción a nivel renal muy tardía, pudiendo encontrarse trazas mas allá de las 48 horas postadministración lo que podría originar fenómenos tóxicos en pacientes susceptibles o con alteración hepática o renal.
MEPIVACAíNA
Metabolizado en un 99% en el hígado con productos mucho menos tóxicos y activos que la lidocaína. Aunque como hemos visto su principal vía de degradación es una vía más saturable que la lidocaína.
PRILOCAINA
En principio un fármaco que parecía ser menos tóxico y más potente que la lidocaína, incluso con mejor tolerancia fetal, sin embargo fue descartado por los anestesiólogos principalmente por ciertas metahemoglobinemias, aunque estas sean una complicación muy infrecuente con un tratamiento rápido mediante la administración de 1 a 2 mg/kg de azul de metileno. Su metabolismo hepático es más rápido que la lidocaína además de permitir una metabolización parcial en ciertos órganos como en el tejido renal y pulmonar. Su excreción renal a nivel tubular específico presenta además un discreto y curioso efecto diurético.
BUPIVACAíNA
Es un fármaco especialmente liposoluble y con una alta fijación a proteínas esto condiciona una resistencia a la hidrólisis del fármaco por parte del organismo. A nivel de excreción podemos decir que la acidificación de la orina puede duplicar su eliminación renal, fenómeno aplicable también a la mepivacaína y a la lidocaína.
ARTICAíNA
Esta amino-amida tiene la particularidad de presentar un enlace ester, lo cual origina una metabolización parcial cuando es hidrolizada por esterasas plasmáticas a metabolitos inactivos. Por ello se dice que su metabolización es muy rápida. Aunque después el organismo deba depurar su metabolito primario, el ácido articaínico, el cual es a su vez es metabolizado posteriormente a glucuronido de ácido articaínico, todos ellos eliminados a través de la orina.
Características intrinsecas y uso clínico
Como hemos dicho al principio de este capitulo la acción de los anestésicos depende en ultima instancia de la cantidad de moléculas activas presentes en el interior axoplásmico de las fibras nerviosas. Dicha cantidad viene dado por múltiples factores dados por la farmacodinámica como por la farmacocinética del fármaco, expondremos brevemente algunos de ellos y las posibles implicaciones en el uso clínico.
Solubilidad y potencia
La correlación entre potencia y solubilidad es bien conocida, particularmente en condiciones in vitro. Es la liposolubilidad del fármaco la que condiciona la facilidad de atravesar los tejidos perineurales y las membranas neuronales. Por ello la bupivacaína racémica y la levobupivacaína son fármacos algo más potentes que la ropivacaína y de 3 a 4 veces más que la lidocaína. Sin embargo a nivel más global sabemos que esta correlación entre liposolubilidad y potencia no es una relación lineal y que cuando el coeficiente de partición lipídica es mayor de 4 no se observa un aumento de potencia en la nueva molécula. Este efecto meseta se debe en parte al atrapamiento del fármaco por las estructuras adyacentes que captan dicho fármaco (grasa especialmente). Este hecho aunque conocido hace mucho tiempo esta siendo estudiado por diversos autores en sitios de administración aún muy inexplorados como por ejemplo el espacio epidural donde la distribución y concentración de la grasa epidural podría ser una de las causas de la variabilidad del efecto de los anestésicos a dicho nivel.
Pka y latencia
El inicio de acción está muy condicionado por el pKa de cada fármaco. Como se explicó en el primer capítulo el porcentaje de porción no ionizada es inversamente proporcional al pKa del anestésico. Por tanto cuanto más alto sea ese valor mayor distancia deberá recorrer el pH de anestésico para que aparezcan las formas no ionizadas liposolubles capaces de atravesar las membranas lipídicas. Es por tanto el principal indicador que nos marca el inicio de acción del anestésico. Este pKa se ve influenciado por factores como la temperatura por lo que sí pueden existir diferencias constatables cuando la conservación de los anestésicos se realiza a diferentes temperaturas.
Toxicidad, concentración y periodo de latencia
La concentración presentada de los anestésicos viene muy determinada por la toxicidad del mismo, por ello generalmente las presentaciones más concentradas son las que contienen el anestésico con mayores márgenes de seguridad. Pudiendo administrar una masa de fármaco mayor que originará una mayor disponibilidad de moléculas capaces de cumplir su función y por tanto un inicio de acción más rápido. Esta premisa supone un punto de partida que algunos autores no asumen pues todavía existe un debate entre volumen / concentración / masa de fármaco como variable fundamental a la hora de valorar la actividad clínica de un anestésico local.
Capacidad de fijación a proteínas y toxicidad
Los anestésicos locales están unidos en gran medida a proteínas, ya sean tisulares o plasmáticas siendo estos conglomerados formas no activas farmacológicamente. Las proteínas principalmente implicadas son por un lado la albúmina como proteína con poca afinidad pero con gran capacidad de fijación por su volumen total en el organismo. Por otro lado están las alfa glicoproteinas que presentan una alta afinidad por estos anestésicos pero en cambio presentan poca capacidad de fijación total por su pequeño volumen de tal manera que son las primeras en «saturarse“.
Existe por tanto una correlación entre capacidad de fijación a proteínas de las moléculas anestésicas y la duración anestésica, esto se explica en parte porque dicha capacidad de fijación determina el porcentaje de la forma ionizada libre y de la no ionizada y por tanto de su efecto. Los conocimientos a nivel molecular ponen de relieve que existen semejanzas estructurales entre la secuencia aminoácida producida por la unión anestésico a la zona del canal iónico donde se une y la conformación creada por el anestésico-proteína plasmática.
Aparte de la duración efectiva del anestésico, la cualidad de fijarse a proteínas condiciona que en neonatos, embarazadas y en pacientes con estados hipoproteicos se pueden originar episodios de toxicidad por disminución de la reserva unida a proteínas plasmáticas y por tanto aumento de las formas activas. Debemos pensar en esos estados hipoproteicos siempre que valoremos tanto dosis administrada como tiempo de administración.
pH y toxicidad
También el pH del organismo condiciona el porcentaje de fijación a proteínas de los anestésicos.
Situaciones de acidosis producen una disminución marcada de dicha fijación del anestésico a las proteínas plasmáticas y por tanto provocan un aumento de la fracción libre del fármaco, de tal manera que podemos encontrarnos con situaciones de toxicidad aunque la cantidad de fármaco en sangre no son en sí tóxicas. Este hecho es especialmente relevante con la bupivacaína cuya concentración de fármaco libre puede aumentar del 5 al 30% solo por el hecho de presentarse una acidosis.
Debemos tener muy en cuenta por tanto la posibilidad de originar una acidosis respiratoria en diversas situaciones, especialmente cuando manejamos sedaciones prolongadas con ventilación espontánea donde la disminución del volumen respiratorio puede desencadenar niveles muy altos de hipercapnia y por tanto originar dicha acidosis respiratoria . Estas situaciones podrían perfectamente hacernos disminuir el margen de seguridad frente a las dosis casi subtóxicas que a veces se aplican en pacientes para infiltraciones de partes blandas en amplias superficies, (por ejemplo cirugía estética). Recordemos que anestésicos como la lidocaína tienen metabolitos activos durante mucho tiempo y situaciones de hipercapnia bien podrían condicionar respuestas deletéreas a nivel de sistema nervioso central aún administrando dosis seguras en administraciones repetidas.
Absorción y toxicidad
Como premisa diremos que la toxicidad de los anestésicos locales aparece cuando el fármaco llega a lugares de acción que no buscamos y para ello debe distribuirse primero por todo el organismo con el fin de originar esos bloqueos de conducción no buscados a nivel cardiaco y de sistema nervioso central principalmente, lugares donde la apertura de los canales se realiza interrumpidamente por ser membranas en continua excitación y por tanto lugares donde el bloqueo fásico se realiza con mayor facilidad.
Ciertos fármacos presentan una acción vasoconstrictora intrínseca que condiciona una menor absorción al torrente circulatorio y por tanto una distribución más lenta hacia zonas que generarían esas acciones de toxicidad sistémica.
La cocaína es un anestésico local con propiedades principalmente vasoconstrictoras intrínsecas, en cambio la mayoría de los anestésicos locales tienen una acción bifásica sobre el músculo liso vascular. A efectos prácticos, a las concentraciones usadas en la clínica habitual se produce un cierto grado de dilatación vascular que condiciona una mayor absorción y distribución por el organismo aunque esa característica no es igual en todos los anestésicos locales.
Para clasificar esa capacidad vasodilatadora exponemos diferentes anestésicos en un orden que sería el siguiente;
Procaína> prilocaína>lidocaína> mepivacaína>bupivacaína racémica
Es llamativo señalar que las formas S o levógiras tienen un efecto vasodilatador mínimo (incluso algún autor refiere cierto efecto vasoconstrictor intrínseco a la levobupivacaína) Lo cual sería una de las causas del mejor perfil de seguridad que tienen estos fármacos aunque como hemos visto antes no es la única.
Así mismo el sitio de administración tendrá una repercusión clara en las posibles toxicidades puesto que cada tejido tendrá una específica vascularización que condicionará un grado distinto de absorción hacia el torrente circulatorio. A modo de recordatorio diremos que los mayores niveles plasmáticos tras una única dosis en diferentes tejidos habituales para la administración de anestésicos se obtienen según este orden.
Traqueal>pleural>intercostal>caudal>paracervical>epidural>braquial>subcutanea>intradural.
Por lo que tendremos que tener en cuenta tal disposición a la hora de valorar dosis y concentración de anestésico para no tener efectos tóxicos en nuestros pacientes pues por ejemplo la dosis administrada a nivel de la mucosa traqueal tiene una biodisponibilidad muy parecida a si hubiéramos administrado el anestésico de manera intravenosa.
Paso placentario
A raíz de los nuevos enantiómeros se ha estudiado el perfil de seguridad a nivel fetal. En estudios con animales se ha visto que estos fármacos atraviesan la barrera placentaria con relativa facilidad.
Sin embargo a modo de crítica constructiva debemos mirar detenidamente estos estudios realizados en animales, ya que su fin es valorar principalmente el paso placentario y no la seguridad fetal. Una visión rápida de ellos nos podría dar una falsa información de qué anestésicos son los más seguros para la anestesia obstétrica pues estos estudios están valorando exclusivamente la facilidad de paso placentario y olvidando aspectos tan relevantes como la metabolización fetal, el porcentaje de fijación a proteínas… etc. en definitiva una valoración completa del perfil de seguridad.
Por ello, desde nuestro convencimiento personal creemos que hoy en día es la levopubivacaína la que presenta un perfil de seguridad fetal mayor en cuanto a su relación de potencia /concentración administrada, a su capacidad de fijación a proteínas y por tanto el porcentaje de fracción libre todo ello configura unos menores efectos a nivel cardiaco y de sistema nervioso central fetal.
pH y taquifilaxia
Especialmente con la lidocaína, podemos observar como puede existir una tolerancia relativamente rápida al anestésico administrado, de tal manera que idénticas dosis repetidas producen cada vez un menor efecto clínico. Esto sucede con menor frecuencia en fármacos con un periodo de acción más largo.
La causa de esto se podría explicar por el pH relativamente bajo de estas soluciones en un medio que tiene un limite al tamponamiento como es el espacio epidural, lo cual origina un aumento de la fracción libre no ionizada incapaz de atravesar las estructuras lipídicas. Sin embargo diversos autores alternativamente proponen que la taquifilaxia puede ser debida también a una modificación de las vías sinápticas centrales de modulación del dolor, de todos modos es práctica habitual el cambiar de anestésico cuando observamos este fenómeno a uno con un pH de conservación mayor, incluso administrando una pequeña dosis de bicarbonato a la mezcla.
Cuadros de los principales anestésicos locales
Aconsejamos encarecidamente al lector que acuda a leer las fichas técnicas de los medicamentos que use, puestos a disposición del público en la página Web de la agencia del medicamento española.
PROCAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD relativa A pH 7,4 | pKa |
| Novocaina | 5,8 | 1 Debil | 8,9 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios | |
| 10mg/kg | 1% uso infiltración | Metabolizado muy rapidamente |
La procaína es un anestésico en desuso actualmente. Sin embargo es un fármaco extremadamente efectivo y todavía se usa para el manejo del espasmo intrarterial para canalización de vías arteriales difíciles. Es así mismo un agente de diagnostico en dolor crónico ideal puesto que la duración del bloqueo de prueba previo a un bloqueo con neurolisis es de tan solo 15 o 45 minutos a lo sumo. Tras la aparición de trastornos neurológicos asociados a la lidocaína parece que hubo un intento de relanzar este anestésico de forma intradural pero su uso se generalizó, máxime cuando parece existir un mayor índice de nauseas cuando se usa este fármaco por esa vía.
Algunos autores aconsejan su uso en el manejo de quemados para retirada de vendajes en una concentración del 1% en suero fisiológico.
La procaína disminuye la contractibilidad del músculo ventricular y atrial y fue usado como antiarrítmico, pero su corta duración hizo desarrollar su análogo; la procainamida.
Es usado actualmente junto a la penicilina que forma una forma menos soluble y por tanto una forma de liberación lenta además de mitigar el dolor a la inyección intramuscular.
TETRACAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Pontocaina, | 75,6 | 200 Elevada | 8,5 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios | |
| 1,5mg/kg | 0,5% caudal / peridural1% raquianestesia | ||
La tetracaina es usada actualmente en geles y colirios para anestesia local o tópica por sus características; es potente, efectiva pero muy tóxica por lo que su uso en mucosas especialmente irrigadas debe ser tenido en cuenta.
LIDOCAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Xilocaina | 64,3 | 150 media | 7,9 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios | Presentación pH |
| 4 mg/kg | 1-2% para intradural,epidural o bloq periférico | presentacion | 6 en presentación 2% |
Esta aminoaciclamidas supuso un hito en la práctica anestésica cuando fue sintetizada por Lofgren en 1943, siendo el prototipo del grupo de las aminoamidas.
Aunque se considera el anestésico local más seguro, pues incluso se usa como antiarrítmico y coadyuvante anestésico de manera intravenosa no hay que olvidar que es según la literatura médica el anestésico local que más muertes provoca por sobredosificación en el mundo puesto de manifiesto por la editorial Albright en 1975 que originó un intenso debate y fue el punto de partida para ofrecer los nuevos enantiómeros al mercado. Presenta un elevado pH de 6 en su concentración al 2% lo cual puede ser de utilidad cuando queremos“ tamponar“ lugares de administración.
PRILOCAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Citanest | 55 | 50 media | 7,9 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios | |
| 6 mg/kg | 1% bloqueo periferico2% intradural y epidural | El más rapidamente metabolizado.Sobredosificación puede dar metahemoglobinemia | |
Un fármaco con poco predicamento en nuestro medio aunque posee unas características muy interesantes. Por un lado es un fármaco de muy fácil metabolización, con unos parámetros hemodinámicos cuando se usa intraduralmente superiores a los de otros anestésicos por lo que su presentación hiperbara la hace recomendable para anestesia intradural en cirugía de corto ingreso, sin embargo su pertenencia a la familia de anestésicos tipo ester sumado a las posibles metahemobloginemias que pueden darse con grandes dosis han supuesto su defenestración en el mercado actual.
MEPIVACAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Carbocaina | 77,5 | 0,8 | 7,6 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios | |
| 3 mg/kg | 1-2% | ||
Su uso en otros países como en el Reino Unido se circunscribe a la odontología. En nuestro medio tiene una utilidad en anestesia intradural de corta duración empleándose al 2% con un volumen entre 2-4 cc para cirugías que no requieren ingreso.
En España es el anestésico de uso preferente para la anestesia local cuando la administra un especialista no anestesiólogo.
BUPIVACAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Marciana | 95,6 | 1000 elevada | 8,1 |
| Dosis única máxima | Concentraciones y uso clínico | comentarios | |
| 3 mg/kg | 0,25 |
Este anestésico presenta el más bajo coeficiente de disociación fármaco-receptor por lo que se considera un fármaco muy letal cuando se alcanzan dosis tóxicas. Actualmente compuestos lipídicos intravenosos se presentan como la única alternativa cuando aparecen fenómenos tóxicos a nivel cardiaco principalmente. A pesar de ello, debido a la cantidad de presentaciones y su flexibilidad de manejo es el anestésico más utilizado en el ámbito de la anestesiología.
LEVOBUPIVACAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Chirocane© | 95,6 – 97% | 1000 elevada (1.624) | 8,1 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios | |
| 3 mg/kg | 0,25 | Es la forma depurada levogira de la bupivacaina |
Este enantiómero levo de la bupivacaína comparte muchas semejanzas con la bupivacaína racémica y algunas diferencias que la hacen notable. Por un lado tiene el mismo pKa 8.1 (8.09 para ser más exactos) un valor que comparte con la ropivacaína y la bupivacaína racémica. Su coeficiente de partición (aceite / agua) es algo mayor, siendo de 1.624 frente a 1.565 de la bupivacaína racémica. Se presenta a pH de 4,0 a 6,5 conteniendo cloruro sódico y agua para la inyección y un ajuste de pH con hidróxido sódico y ácido clorhídrico. Se debe preservar de la luz y conservar a 30 º.
Respecto a la bupivacaína racémica presenta un mayor grado de unión a proteínas plasmáticas (mayor del 97% según algunos autores) con un volumen de distribución menor, mayor aclaramiento plasmático y una vida media algo más corta. Su toxicidad es en estudios con animales a dosis letales hasta dos veces menores.
Actualmente es el fármaco que presenta un perfil de seguridad más alto en cuanto potencia/dosis letal/duración, muy aproximado al de la ropivacaína aunque la potencia menor de esta influye negativamente en dicho perfil de seguridad cuando la comparamos con la levobupivacaína.
ROPIVACAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Naropin© | 94 | 400 elevada | 8,1 |
| Dosis única maxima | Concentraciones y uso clínico | comentarios |
Comercializada en 1.997 en España supuso un hito en la seguridad de estos fármacos en cuanto fue la primera forma depurada de un enantiómero. De gran predicamento en la anestesia obstétrica, ha sido profusamente estudiada y comparada con otros fármacos, incluso con la llegada de la levobupivacaína mediante estudios que a veces adolecían de errores de planificación y originaban hipótesis contradictorias con las bases farmacológicas de los fármacos estudiados.
Su uso a nivel intrarraquideo ha sido no aceptado de manera oficial hasta hace relativamente poco, aceptándose su uso en base a la evidencia de seguridad clínica que han ofrecido estudios hechos por autores que no han tenido en cuenta la ficha técnica del medicamento.
ARTICAINA
| SINONIMOS | % FIJACION PROT | LIPOSOLUBILIDAD A pH 7,4 | pKa |
| Articain © | 7.8 | ||
| Dosis única maxima5mg/kg | Concentraciones y uso clínico | comentarios |
Anestésico local tipo amida.
Potencia de 1.5 la de la lidocaína y 1.9 la de procaína.
Toxicidad similar a lidocaína y procaína.
Pka 7.8
Ph en solución simple no disponible; en solución con vasoconstrictor: 1:100000 4.4.5.2
1:200000 4.6-5.4
Metabolismo: Es el único anestésico local tipo amida que contiene un grupo tiofeno, además de un grupo éster por lo que su metabolización se produce tanto por hidrólisis por esterasas plasmáticas, como por enzimas microsomales hepáticas.
Su metabolito principal que es el ácido articaínico es inactivo desde el punto de vista farmacológico.
Semivida anestésica de 0.5 horas.
Excreción vía renal, un 5-10% sin metabolizar y un 90% metabolizado ( M1 87% y M2 2%).
Dosis máxima en adultos de 7 mg/kg (500mg Máximo absoluto) y en niños entre 4-12 años no superar los 5 mg/kg, en el resto no se debe exceder los 7 mg/kg; no usarse en menores de 4 años.
Comienzo de acción: 1:100000 en infiltración 1-2 min (duración aprox 60-75 min) y en bloqueo mandibular 2-3 min( duración aprox 180-360 min); 1:200000 en infiltración 1-2 min (duración aprox 45-60 min) y 2-2.5 min para bloqueo mandibular (duración aprox 120-300).
Posee propiedades fisicoquímicas como otros anestésicos locales con excepción de molécula aromática y el grado de unión a proteínas.
Difunde a partes blandas por lo que puede anestesiar en paladar blando sin necesidad de infiltrarlo.
Se ha relacionado con parestesias a concentraciones del 4%.
Se registraron algunos casos de metahemoglobinemia en relación con su uso a altas dosis por vía IV con fines de anestesia regional.
Contraindicaciones absolutas en pacientes con sensibilidad a anestésicos tipo amida y/o al sulfito.
Al principio los cartuchos que se comercializaban tenían como conservante el metilparaben, con el consiguiente riesgo de alergias, pero en las presentaciones actuales ya se ha retirado.
Nombres comerciales:
1:100000 Septocaine, Septanest SP, Astracaine, Ultracaine D-S forte.
1:200000 Septanest S, Astracaine, Ultracaine.
Todos con una concentración del 4%.
Bibliografía
1.- Comparison of 2% lignocaine with adrenaline and fentanyl, 0.75% ropivacaine and 0.5% levobupivacaine for extension of epidural analgesia for urgent caesarean section after low dose epidural infusion during labour.Anaesth Intensive Care. 2008 Sep;36(5):659-64.Sng BL, Pay LL, Sia AT. (PubMed)
2.- A comparison of the inhibitory effects of bupivacaine and levobupivacaine on isolated human pregnant myometrium contractility Fanning RA, Campion DP, Collins CB, Keely S, Briggs LP, O’Connor JJ, Carey MF.Anesth Analg. 2008 Oct;107(4):1303-7. (PubMed) (Pdf)
3.- Bupivacaine, levobupivacaine and ropivacaine: are they clinically diferent?“. Andrea Casati MD, Marta Putzu MD. Best practice & Research Clinical Anesthesiology. June 2005, vol 19, issue 2;p 247-268. (PubMed)
4.- A randomized sequential allocation study to determine the minimum effective analgesic concentration of levobupivacaine and ropivacaine in patients receiving epidural analgesia for labor. Dan Benhamou, MD., Caroline Ghosh MD., Fréderic J Mercier, MD., Ph.D. Anesthesiology 2003;99:1383-6. (PubMed)
5.- The relative motor blocking potencies of bupivacaine and levobupivacaine in labor. Hector J. Lacassie, MD; Malachy O. Columb, F.R.C.A. Anesth Analg 2003;97:150. (PubMed) (Pdf)
6.- Relative analgesic potencies of levobupivacaine and ropivacaine for epidural analgesia in labor. Linda S. Polley, MD;Malachy O.Columb F.R.C.A. Anesthesiology 2.003;99:1354-8. (PubMed)
Para saber más
– Aurelio Gomez Luque. Puesta al día en anestesia regional y tratamiento del dolor. Volumen II 2.007 .
– Anestesia Locorregional. P.Gauthier – Lafaye. 1987 .Ed Masson 1.985.
– Principles and Practice of Pharmacology for Anesthetists. 5ª ed 2008. TN Calvey and NE. Williams.
– Farmacología de los anestesicos Locales. J.M. de Carlos y MA Viamonte. 1.999.
– Medicamentos habitualmente empleados para el bloqueo nervioso. Farmacología de los anestésicos locales de Cosmo A. Cap 39 de Tratamiento practico del dolor .P. Prithvi Raj ed Harcout 2.002.
Dr A.Gironés Muriel. H. Sanitas La Moraleja. Madrid. España.Dr. Angel Villar-Pellit. H Virgen de la victoria .Málaga. España.

Buenas noches , por favor si pudiera enviarme alguna revision sobre predictores de via aerea, y manejo de via aerea dificil. gracias.
Hola Jesús Padrón:
Soy Marisa Mariscal anestesista del Hospital de Getafe . Madrid.
Muy interesada en los temas de Vía Aérea Dificil, si te interesa, hemos escritos varios temas entre ellos predicción en VAD en la página www. arydol.es y también publicamos en 2008 un libro sobre el tema: «Manejo actual de la vía aérea dificil» M. L . Mariscal Flores, M.L. Pindado Martinez, lo puedes encontrar poniendo el título en Google.
Espero haberte ayudado.
Un saludo. Marisa Mariscal
Hola..quisiera saber si alguien me puede ayudar con algo de monitoreo renal y del sistema nervioso central???
Hola:
Me gustaría conocer el resultado positivo o no de combinar dos anestésicos locales tales como mepivacaína al 2% y ropivacaína al 0,75% en bloqueos nerviosos periféricos o anestesia loco-regional.
Muchas gracias.
Soy odontologa y quisiera saber si cual de los embases de vidrio o de plastico es mejor para los tubitos de anestesicos dentales y porque?
EN GENERAL SON MEJORES LOS DE VIDRIO SOBRE TODO CUANDO EL ANÉSTESICO EN CUESTIÓN TIENE ADRENALINA / EPINEPHRINA.
ESTO ES DEBIDO A QUE LA ADRENALINA ES MUY SENSIBLE AL OXíGENO Y EL PLíSTICO ES PERMEABLE AL OXíGENO MIENTRAS QUE EL VIDRIO NO LO ES
VISTO BUENO PARA REALIZAR TERAPIA NEURAL CON MEPIVACAíNA CON RESULTADOS EXCELENTES EN PACIENTES EN GRADO NEURíLGICO AVANZADO DE PARíLISIS FACIAL GRADO 3
Buenos días
Soy odontóloga,y quisiera preguntarle que anestésico aconsejaría para uso odontologico en mujeres embarazadas,teniendo en cuenta que los preparados en carpules que se comercializan son a base de lidocaína,mepivacaína o articaína.
Muchas gracias
Muy interesante el artículo!
TOod ha cambiado a la anestecia dental se le agrega adrenalina y epenifrina para alargar su duración el cuerpo con estoas los aodntologos ya no necesitan tener manos de seda ya no te hacen doler porque esta anestecia es muy fuerte