Roca Viéitez O (1), Bernaldo de Quirós de Cal B (2), Díaz Mosquera L (1)
1 Servicio de anestesia, reanimación y terapéutica del dolor. Complejo Hospitalario Universitario de A Coruña.
2 Servicio de anestesia, reanimación y terapéutica del dolor. Complejo Hospitalario Universitario de Ferrol.
INTRODUCCIÓN
El uso de fármacos anticonvulsivantes es una constante durante el día a día de nuestra labor como anestesiólogos, enfrentándonos a pacientes epilépticos que acuden a intervenirse o convulsiones que suceden durante el perioperatorio. Existen multitud de fármacos antiepilépticos (FAEs) pero únicamente unos pocos están disponibles para su administración oral. A lo largo de este artículo nuestro objetivo es realizar una revisión de la literatura publicada para condensar la información obtenida y hacer un breve resumen actualizado de los conocimientos que debe tener todo anestesiólogo sobre este tipo de fármacos.
HISTORIA
Una convulsión es el síntoma resultado de la actividad neuronal anómala excesiva o simultánea en el cerebro. Es importante recordar que convulsión no implica epilepsia. La búsqueda de remedios para estas convulsiones han sido una constante a lo largo de la historia.
Ya los incas y los aztecas hacen referencia a ellas, achacándolas a la acción de espíritus malignos y basando el tratamiento en métodos mágicos y en el empleo de brebajes a base de plantas, sangre de animales como el perro o el puma y el polvillo del rascado de las piedras. Avicena, uno de los médicos más antiguos de los que se tiene constancia describía la terapia dietética como una de las más eficaces (abstinencia de zanahoria, cebollas, productos derivados de la leche, frutas secas, pescado y vino). El Doctor Broca (que da nombre a la afasia de Broca), describió como durante una larga época en la antigüedad se empleaba la trepanación como vía de escape de los espíritus diabólicos. El comienzo de la terapia farmacológica se retrasó hasta en la segunda mitad del siglo XIX (1857) con el descubrimiento de las propiedades antiepilépticas de los bromuros que fueron el principal tratamiento hasta que el descubrimiento del fenobarbital en 1912. Desde entonces han aparecido de forma progresiva múltiples fármacos con efecto anticonvulsivo.
MECANISMO DE ACCIÓN
El efecto de los fármacos antiepilépticos en la supresión de las crisis epilépticas está relacionado con la acción sobre diferentes dianas disminuyendo la excitabilidad neuronal y la hipersincronía de los circuitos cerebrales. La mayoría de los FAE presenta diferentes mecanismos de acción. De forma resumida los principales mecanismos de acción son:
1. Acción principal sobre los canales iónicos voltajes dependientes.
- Bloquear o modular canal de sodio (el más común): se reducen las descargas neuronales repetitivas rápidas, estabilizando la membrana neuronal, disminuyendo la actividad epiléptica y la progresión de las crisis.
- Bloquear canales de calcio. Existen tres tipos de canales de calcio en las neuronas, que se distinguen por su tasa de reactivación y el voltaje.
- La gabapentina y la pregabalina se unen a la proteína α2δ auxiliar de un canal de calcio dependiente de voltaje y pueden reducir la despolarización mediada por calcio al inhibir las corrientes de calcio hacia el interior y reducir la liberación de neurotransmisores excitatorios.
- Apertura de los canales de potasio: facilita el restablecimiento del potencial de reposo.
2. Acción principal sobre el sistema GABA (agonista). El ácido gamma-aminobutírico (GABA) es un neurotransmisor que se distribuye ampliamente por todo el sistema nervioso central y ejerce una inhibición postsináptica. De hecho, el mecanismo de acción de algunos proconvulsivos consiste en unirse al receptor GABA evitando la inhibición postsináptica.
- El tono GABAérgico reducido se considera proconvulsivo.
- El aumento del tono GABAérgico generalmente tiene un efecto anticonvulsivo.
La síntesis de GABA depende de la enzima ácido glutámico descarboxilasa (GAD), que requiere piridoxina como coenzima (por eso los bebés con déficit de la misma convulsionan) generando GABA a partir de la descarboxilación de glutamato. Después GABA, se metaboliza a succinato por medio de la enzima mitocondrial GABA transaminasa (GABA-T).
Por lo tanto, es fácilmente deducible que los antiepilépticos buscan aumentar el suministro de GABA mediante la reducción del metabolismo de la misma por GABA-T (vigabatrina), el aumento de su producción por GAD o reducir la recaptación de GABA en las neuronas (tiagabina). Se han diseñado otros medicamentos anticonvulsivos para imitar la acción del GABA, mientras que otros mejoran la inhibición mediada por el GABA endógeno. Benzodiacepinas y fenobarbital se unen al recetor A de GABA facilitando la unión de dicho receptor a GABA.
3. Acción principal sobre los receptores ionotrópicos de glutamato (receptores AMPA, kainato, NMDA, glicina). Disminuyen su actividad (antagonismo).
El glutamato es el neurotransmisor excitador más prevalente. Hay dos tipos de receptores de glutamato: ionotrópicos, que forman canales iónicos que se activan por la unión del glutamato y que se cree que tienen un papel en la generación y propagación de convulsiones, y metabotrópicos, que activan indirectamente los canales iónicos a través de la cascada de señalización de la proteína G.
4. Acción principal sobre la modulación de la maquinaria que facilita la liberación sináptica de neurotransmisores.
- Levetiracetam y Brivaracetam se fijan a la proteína SV2A, localizada en las vesículas presinápticas, facilitando la liberación de neurotransmisores inhibitorios.
INTERACCIONES FARMACOCINÉTICA y FARMACODINÁMICAS
Podemos diferenciar dos grandes tipos de interacciones:
- Farmacocinéticas: relacionadas con los procesos a que se ven sometidos los fármacos en su paso por el organismo, desde que se administra hasta su eliminación.
- Farmacodinámicas: relacionadas con su actividad biológica, mediatizada por el mecanismo de acción.
Interacciones farmacocinéticas
Absorción: Se absorben bien por vía digestiva. La gabapentina tiene una absorción saturable, de modo que, por encima de 1.200 mg/día, su absorción se reduce en un 10-35 %.
La presencia de comida retrasa la absorción de muchos FAE, pero no modifica la cantidad total de fármaco absorbida. La administración conjunta con comida permite disminuir los efectos secundarios asociados al pico de dosis sin variar la eficacia de la medicación.
Los antiácidos pueden alterar la absorción fundamentalmente de la fenitoína y, aún en mayor medida, de la gabapentina.
Distribución:
Los fármacos más liposolubles tienden a circular en la sangre unidos a proteínas plasmáticas, sobre todo, la albúmina. Si coinciden varios FAE con alta afinidad por proteínas plasmáticas, pueden competir y desplazarse unos a otros, lo que aumentaría la fracción libre, a la que va ligada la eficacia y los efectos secundarios. Por lo tanto, debemos tener cuidado en situaciones que suelen acompañarse de hipoproteinemia, como la desnutrición o la edad avanzada.
Transporte a través de la barrera hematoencefálica:
El paso a través de barreras, tanto digestiva como hematoencefálica, está regulado por una serie de proteínas transportadoras, como la glicoproteína P, que tienen una acción detoxificarte, extrayendo moléculas extrañas, como los fármacos, del sistema nervioso central.
Los FAE inductores enzimáticos potentes también incrementan la expresión de estas proteínas transportadoras en los distintos epitelios, con lo que no sólo modifican el metabolismo, sino también la absorción y penetración cerebral de los fármacos que son sustratos de estas.
Metabolismo:
Carbamacepina, fenitoína, fenobarbital y primidonason potentes inductores de múltiples subunidades de los sistemas enzimáticos CYP450, UGT y epóxido-hidrolasa. En consecuencia, aceleran el metabolismo y disminuyen los niveles plasmáticos de todos los fármacos y sustancias orgánicas (vitaminas y hormonas) que se metabolizan por estas vías.
Oxcarbazepina y eslicarbazepina tienen una acción inductora intermedia, que se contrarresta en parte porque inhiben otras subunidades de CYP450.
Topiramato y perampanel son inductores débiles, con una acción sólo evidente a dosis muy elevadas.
Resumiendo, el uso de FAE inductores potentes puede reducir los niveles plasmáticos y disminuir la eficacia de otros fármacos de metabolismo hepático, incluyendo a ellos mismos. Por ejemplo, es típico ver como el incremento progresivo de dosis de carbamazepina no causa un aumento proporcional de sus niveles plasmáticos, debido a la autoinducción de su metabolismo. Así que en general cuando se combina un FAE de metabolismo hepático con un inductor potente, es necesario utilizar dosis más elevadas que cuando se combina con un inductor débil o con un no inductor. También, la vida media del fármaco inducible se acorta, en fármacos de vida media más corta puede ser importante y obligar a un incremento en el número de tomas, para evitar periodos de infradosificación entre una dosis y otra.
La supresión de FAE inductores lleva aparejada un incremento en la concentración, y en los efectos secundarios, provocados por otros fármacos que pueda estar tomando el paciente. Al contrario de lo que ocurre con la inducción, la inhibición es inmediata. El ácido valproico es el inhibidor más potente. Cuando se añade un FAE de metabolismo hepático, como lamotrigina o Carbamacepina, a un paciente que toma ácido valproico, es recomendable comenzar con una dosis más baja y realizar una escalada más lenta, para evitar efectos indeseables.
Interacciones farmacodinámicas
Lo fundamental es entender que el objetivo es encontrar combinaciones de FAEs sinérgicas, es decir, que la combinación de los dos fármacos obtenga una acción superior a la de cada uno de ellos por separado. En general, se consigue con fármacos con diferente mecanismo de acción. Lo ideal es conseguir una combinación sinérgica en eficacia y antagonista en efectos adversos. La más conocida es la sinergia ácido valproico con lamotrigina.
EFECTOS ADVERSOS
El mecanismo de acción de los FAE no es selectivo y así actúan sobre redes o grupos neuronales “normales”. Es por ello por lo que la mayoría de los FAE actuales provocan toxicidad neuronal como efecto secundario común. Obviamente, el impacto clínico y la frecuencia de la presentación de los efectos secundarios depende de múltiples condicionantes (edad, sexo, comorbilidades.). En general podemos encontrarnos con:
Efectos adversos dosis-dependientes (tipo A). Los más frecuentes. Suelen presentarse de forma aguda al inicio y, con el tiempo, pueden disminuir y ser tolerados. Desaparecen con la reducción de la dosis y es inhabitual que obliguen a la retirada del FAE., incluso en ocasiones se puede llegar a tolerar una dosis inicialmente no tolerada realizando un escalado más lento Dependiendo del fármaco hay descritos unos u otros, pero grosso modo pueden ser de dos tipos:
- Sistémicos: náuseas, vómitos, estreñimiento, sensación de fatiga, hiponatremia (SIADH), erupción cutánea.
- Neurológicos: temblor, mareo, ataxia, irritabilidad, somnolencia.
Efectos adversos idiosincráticos (tipo B): son menos frecuentes e impredecibles. Se producen por diferentes vías: reacciones de hipersensibilidad, interacción con órganos diana erróneos o por citotoxicidad directa del FAE o de sus metabolitos.
- Efectos hematológicos (leucopenia, neutropenia).
- Broncoespasmo (típico del BRV)
- Psicosis. Ideación suicida.
- Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica.
- Síndrome de hipersensibilidad a los FAE.
- 1/10.000 pacientes expuestos.
- Fiebre, rash cutáneo, linfadenopatías y fracaso multisistémico.
- Mortalidad del 50 %.
- El tratamiento consiste en la retirada del FAE y adición de corticoides e inmunoglobulinas intravenosas.
Efectos adversos crónicos (tipo C): curso insidioso, mala tolerabilidad y pueden aparecer tras exposición prolongada, son causa de retirada:
Efectos sobre la cognición, antiestéticos (alopecia, ganancia ponderal, hiperplasia gingival, hirsutismo e hiperpigmentación de mucosas), endocrinológicos (déficit de vitamina D, osteomalacia, disfunción sexual, hipotiroidismo), urológicos (litiasis renal) y visuales (hiperpigmentación retiniana, reducción del campo visual).
Efectos teratógenos y carcinogénicos: los efectos carcinogénicos se han relacionado con fenobarbital y fenitoína en modelos animales. El tratamiento prolongado con fenitoína se relaciona con pseudolinfoma (simula un linfoma y se resuelve con la suspensión del FAE).
¿CÓMO EMPLEARLOS?
En general se acepta que la vía oral es la de elección incluso en pacientes hospitalizados. Sin embargo, en ocasiones no resulta posible por el estado general del paciente o por otras circunstancias. En estos casos podemos utilizar la vía intravenosa, sin embargo, es reducido el número de fármacos disponibles con esta presentación en comparación con el gran arsenal terapéutico de antiepilépticos por vía oral. Los más utilizados son el ácido valproico, el levetiracetam y la lacosamida, cuya equivalencia es la misma entre la vía oral y la intravenosa. La dosis total diaria de ácido valproico debe administrarse en forma de perfusión continua.
Fármacos antiepilépticos en formulación intravenosa:
- Fenobarbital (Luminal): Ampollas 200 mg/1 ml
- Fenitoína (Fenitoína Rubio): Ampollas 100 mg/2 ml y 250 mg/5 ml
- Ácido valproico (Depakine): Vial 400 mg (+ ampolla 4 ml)
- Clonazepam (Rivotril): Ampollas 1 mg/1 ml
- Levetiracetam (Keppra): Vial 100 mg/ml (5 ml)
- Lacosamida (Vimpat): Vial 10 mg/ml (20 ml)
- Brivaracetam (Briviact): Vial 10 mg/ml (5 ml)
La evidencia apoya el inicio en monoterapia en dosis bajas, ya que la mitad de los pacientes logran controlarse de esta forma. En niños en general el metabolismo y eliminación renal están acelerados. Resulta en una semivida más corta, por lo que es habitual el uso de más tomas al día para mantener concentraciones terapéuticas. Globalmente, se requieren dosis por peso mayores en el primer año de vida (cociente absorción/eliminación muy bajo a esta edad).
NIVELES PLASMÁTICOS
Los niveles plasmáticos de un FAE deben solicitarse en período estacionario, situación que aparece tras haber transcurrido 4-5 semividas desde el inicio del tratamiento o último cambio de dosis.
No hay evidencia de beneficio de evaluar rutinariamente niveles plasmáticos de FAE, ya que se ha visto que no aumenta el control de los pacientes ni disminuye los efectos adversos. Según las guías de la Sociedad Española de Neurología estaría indicado en las siguientes situaciones:
- Comprobar el cumplimiento y la adherencia al tratamiento.
- Sospecha de toxicidad, es especial en politerapia donde el responsable de los efectos adversos del paciente puede ser uno o varios fármacos.
- Para definir el intervalo terapéutico individual de un paciente. Se deberían solicitar dos determinaciones separadas en el tiempo dos – cuatro meses para promediar la variabilidad.
- En caso de epilepsia mal controlada, a pesar de monoterapia idónea o politerapia. Permite identificar que modificaciones en la pauta posológica puedes ser más útiles.
- Durante el embarazo.
- Otros grupos poblaciones y estadios fisiopatológicos en los que se prevé distinta farmacocinética (niños, ancianos, cirugía bariátrica, insuficiencia renal o hepática, quemados, pacientes críticos, enfermedad infecciosa, hipoalbuminemia, hemodiálisis etc.).
- Si se producen cambios en la forma farmacéutica o en la especialidad farmacéutica.
- Cuando exista sospecha de interacciones.
- Cuando se ha llegado a una dosis considerada adecuada y el paciente continúa presentando crisis. Los niveles pueden ayudar a valorar si el paciente es un metabolizador rápido o no sigue el tratamiento de forma adecuada.
Además, en aquellos FAE con alta unión a proteínas plasmáticas (fenitoína, carbamazepina, ácido valproico y perampanel) en ocasiones las concentraciones totales del fármaco no permiten estimar la concentración libre, es decir, la que en último término realiza la acción terapéutica. Existen una serie de situaciones donde la la Sociedad Española de Neurología recomienda determinar la fracción libre:
- Pacientes urémicos o con enfermedad renal grave.
- Enfermedad hepática grave
- Albúmina sérica < 25 g/l.
- Hiperlipidemias.
- Tratamiento concomitante con fármacos que presentan también alta unión a proteínas plasmáticas y que, por tanto, pueden competir en su unión.
- Concentraciones séricas totales dentro del intervalo de referencia y presencia de signos o síntomas de toxicidad o pacientes sin respuesta terapéutica adecuada.
PRINCIAPLES ANTICONVULSIONANTES EMPLEADOS EN REANIMACIÓN
Levetiracetam
Mecanismo de acción: el principal mecanismo de acción del levetiracetam es la unión a la proteína vesicular sináptica SV2A. Esto parece resultar en una disminución inespecífica en la liberación de neurotransmisores También parece actuar sobre el sistema GABA.
Indicaciones: fármaco de amplio espectro, eficaz contra las convulsiones focales, convulsiones tónico-clónicas generalizadas y las convulsiones mioclónicas generalizadas (único medicamento anticonvulsivo con evidencia Clase I de eficacia contra las convulsiones mioclónicas). Estatus epiléptico (segunda línea).
Metabolismo e interacciones:
El metabolismo de levetiracetam es independiente del sistema CYP (citocromo P450) –es por hidrólisis enzimática-, lo que limita el potencial de interacción farmacocinética con otros medicamentos (anticonvulsivos, anticonceptivos hormonales o fármacos inmunosupresores comúnmente utilizados en trasplantes de órganos). Algunos estudios han encontrado que la coadministración de medicamentos anticonvulsivos inductores de enzimas se asocia con un aumento de aproximadamente el 25 por ciento en la depuración de levetiracetam, sin embargo se considera que esto tiene una importancia clínica limitada.
Posología (Figura 1 y 2):
Dosis inicial 500 mg dos veces al día. Dosis máximas 4000 mg al día. Sin embargo, no se ha establecido que las dosis superiores a 3000 mg diarios tengan un beneficio adicional y es más probable que causen efectos secundarios. Una dosis de carga con 1500 mg es bien tolerada y produce rápidamente concentraciones séricas terapéuticas. Se recomienda una dosificación basada en el peso para niños menores de 16 años: 10 mg/kg dos veces al día produce niveles que se aproximan a los de la dosis de 500 mg dos veces al día en adultos, con 20 mg/kg dos veces al día como dosis objetivo habitual para concentraciones terapéuticas.
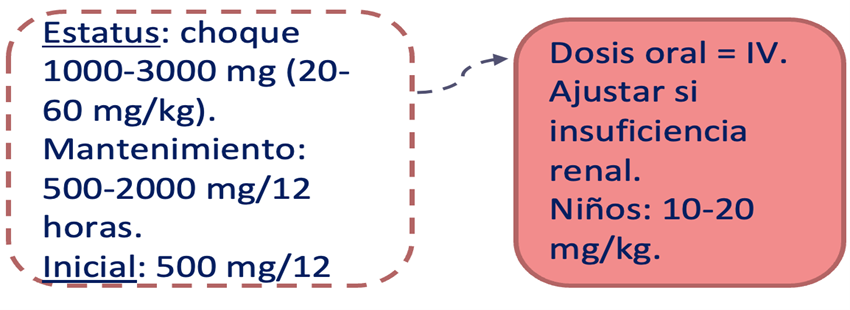
Figura 1. Resumen dosificación Levetiracetam (elaboración propia).

Figura 2. Dosificación de Levetiracetam según aclaramiento de creatinina (elaboración propia).
La formulación IV de levetiracetam está aprobada para su uso en situaciones clínicas cuando los pacientes no pueden tomar medicamentos orales temporalmente. La infusión es bioequivalente a las tabletas orales. Se encuentra disponible una formulación de liberación prolongada de levetiracetam; los estudios sugieren que es efectivo y bien tolerado, se toma una vez al día.
No se requiere un control de rutina de los niveles de levetiracetam. Sin embargo, los niveles séricos pueden ser útiles en ciertas condiciones que se espera que influyan en los niveles de levetiracetam, como el embarazo, la insuficiencia renal y el uso concomitante de fármacos inductores (p. ej., carbamazepina).
Efectos adversos:
- La mayoría de los eventos adversos son de intensidad leve a moderada y ocurren con mayor frecuencia durante la fase de titulación inicial
- Más comunes: fatiga, somnolencia (el más frecuente), mareos e infección de las vías respiratorias superiores (faringitis es característica).
- Los efectos secundarios neuropsiquiátricos (alteración del comportamiento, psicosis, alteración estado de ánimo) razón más común para la interrupción del fármaco.
- En los niños, los problemas de comportamiento y la somnolencia son los efectos secundarios más comunes.
- Algunos informes sugieren que la suplementación con piridoxina puede reducir los efectos adversos neuropsiquiátricos del levetiracetam.
- Otros: trombocitopenia, pérdida de peso.
Brivaracetam:
Mecanismo de acción: igual que el levetiracetam.
Indicaciones: amplio espectro de eficacia en modelos preclínicos, los ensayos de clase I en humanos solo se han realizado en pacientes con convulsiones focales. Sin embargo, los datos abiertos respaldan la eficacia para los tipos de convulsiones generalizadas.
Metabolismo e interacciones:
Se metaboliza principalmente (60 %) por citocromo P450 (CYP) y, de forma secundaria (30 %) a través del CYP2C19 hepático. El gen CYP2C19 es polimórfico y los polimorfismos asociados con la función reducida de CYP2C19 tienen el potencial de disminuir el metabolismo de brivaracetam y, por lo tanto, aumentar la toxicidad. Por lo tanto, el uso concomitante de fenitoína, carbamazepina, fenobarbital y otros inductores de CYP2C19 se asocia con una disminución de las concentraciones plasmáticas de brivaracetam. No es un inductor enzimático como tal, pero se ha visto que aumentar las concentraciones plasmáticas de fenitoína y de la carbamazepina.
Dosificación:
Dosis inicial recomendada por el fabricante para adultos y niños mayores de 16 años es de 50 mg dos veces al día. La dosis puede ajustarse a 25 mg dos veces al día o aumentarse a 100 mg dos veces al día según la respuesta terapéutica y la tolerabilidad. Se recomiendan dosis más bajas para pacientes con insuficiencia hepática.
El fármaco está disponible en formulaciones orales e intravenosas (IV). Cuando la administración oral no es factible temporalmente puede administrarse por vía intravenosa. La dosis y la frecuencia para la preparación IV son las mismas que para las tabletas orales y la solución oral.
Efectos adversos:
Más comunes: somnolencia, mareos, fatiga y náuseas. Reacciones graves de hipersensibilidad, como broncoespasmo y angioedema (muy raras). Efectos psiquiátricos: irritabilidad, ansiedad, insomnio, y depresión.
Bivaracetam frente al levetiracetam:
La tolerabilidad comparativa se desconoce. La afinidad es aproximadamente 20 veces mayor y una mayor selectividad por la proteína. También tiene una mayor permeabilidad cerebral (interesante en el estatus epiléptico). Una revisión sistemática de 2021 identificó cinco estudios que incluían un subconjunto de personas con epilepsia que cambiaron de levetiracetam a brivaracetam, lo que se asoció con una mejora en los eventos adversos conductuales. No es efectivo cuando se agrega a levetiracetam.
Lacosamida:
Mecanismo de acción: inactivación lenta de los canales de sodio dependientes de voltaje. También se une a la proteína 2 mediadora de la respuesta a la colapsina (CRMP2), que puede estar involucrada en la epileptogénesis.
Indicaciones:
La lacosamida es eficaz contra las convulsiones de inicio focal y las convulsiones tónico-clónicas de inicio generalizado. No es eficaz en mioclonías.
Metabolismo e interacciones:
Los inhibidores potentes de CYP (p. ej., valproato) pueden disminuir la eliminación en presencia de insuficiencia hepática o renal.
Dosificación (figura 3):
Oral e IV (aprobada para pacientes ≥4 años de edad). Dosis de 50 mg dos veces al día como terapia adyuvante en adultos; la dosis inicial recomendada como monoterapia es de 100 mg dos veces al día. La dosis puede aumentarse a intervalos semanales en 100 mg al día hasta una dosis de mantenimiento de 200 a 400 mg (total) al día. Se debe ajustar la dosis en pacientes con insuficiencia hepática o renal y se debe complementar después de la hemodiálisis.
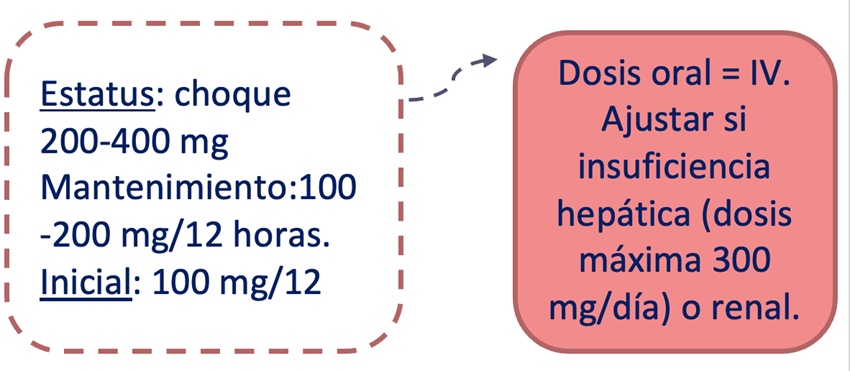
Figura 3. Resumen dosificación de Lacosamida (elaboración propia).
Efectos adversos:
Bien tolerada. Mareos, náuseas, vértigo, coordinación anormal y ataxia son los efectos secundarios más frecuentes. Las prolongaciones del intervalo PR dependientes de la dosis en el electrocardiograma (ECG) en algunos pacientes estudiados sugieren precaución al prescribir lacosamida a aquellos con problemas de conducción conocidos (p. ej., bloqueo auriculoventricular, disfunción del nódulo sinusal sin marcapasos, síndrome de Brugada), cardiopatía isquémica o estructural grave, o uso concomitante de medicamentos que prolongan el intervalo PR. Se han descrito bloqueo auriculoventricular de segundo grado, aleteo/fibrilación auricular y síncope.
Clonazepam:
Mecanismo de acción: sistema GABA.
Metabolismo e interacciones: el clonazepam se metaboliza en el hígado, principalmente por la isoenzima 3A4 del citocromo P450 (CYP3A4), con potencial de interacciones que involucran a otros sustratos de CYP3A4.
Posología: la dosis inicial de clonazepam en adultos es de 0,5 a 1 mg/día cuando se usa como terapia adjunta, o de 0,5 a 1,5 mg/día cuando se usa como monoterapia, con incrementos semanales de 0,5 a 1 mg/día según sea necesario y tolerado (máximo 20 mg diarios). La dosis habitual de mantenimiento es de 2 a 8 mg diarios divididos en una o dos tomas.
Efectos adversos: los efectos adversos del clonazepam incluyen principalmente somnolencia, ataxia y cambios de comportamiento [41].
Benzodiacepinas:
El lorazepam, el diazepam (especialmente el diazepam rectal o nasal) y el midazolam nasal se usan típicamente como medicamentos de rescate para las convulsiones repetitivas agudas o el estado epiléptico. Como clase, las benzodiazepinas pueden estar asociadas con el desarrollo de tolerancia, lo que limita su utilidad en el tratamiento crónico de la epilepsia. La interrupción repentina de las benzodiazepinas puede provocar convulsiones por abstinencia.
Ácido valproico:
Mecanismo de acción: valproato (ácido valproico) tiene múltiples mecanismos de acción. El valproato parece suprimir la descarga neuronal repetitiva de alta frecuencia mediante el bloqueo de los canales de sodio dependientes del voltaje, pero en sitios diferentes a los de la carbamazepina y la fenitoína. El valproato aumenta las concentraciones cerebrales de GABA en dosis clínicamente relevantes. El valproato no parece tener ningún efecto directo sobre el receptor GABA(A), pero la liberación de GABA puede verse potenciada por un efecto presináptico del valproato sobre los receptores GABA(B). La inhibición de la transaminasa GABA terminal nerviosa (GABA-T) probablemente también aumenta los niveles presinápticos de GABA. Además, el valproato puede aumentar la síntesis de GABA al activar la descarboxilasa del ácido glutámico (GAD). Finalmente, el valproato actúa contra las corrientes de calcio tipo T, aunque esta acción es más débil que la observada con etosuximida.
Indicaciones: El valproato es un medicamento anticonvulsivo de amplio espectro que se usa solo y en combinación para el tratamiento de convulsiones generalizadas y focales.
Metabolismo e interacciones: Se une estrechamente a proteínas, por lo que hay que tener cuidado en hipoalbuminemia.Inhibidor del CYP. Requiere ajuste de dosis en insuficiencia hepática.Contraindicado si trastornos conocidos del ciclo de la urea (riesgo hiperamonemia grave).
Dosificación:
Estatus: dosis de choque: 15-45 mg/kg (velocidad a < 6 mg/kg/min). Dosis mantenimiento 1 mg/kg/h o en tres dosis. Ajustar según concentraciones alcanzadas. Si epilepsia previa tratada con ácido valprocico debemos iniciar la administración intravenosa 4-6 horas después de última toma oral. Misma dosis oral que intravenosa.
Efectos adversos:
- Resistencia insulina. Síndrome metabólico. Síndrome ovario poliquísitco.
- Encefalopatía hiperamonémica (encefalopatía aguda o subaguda y convulsiones sin necesidad de anomalías en pruebas de función hepática: suspender valproico, carnitina y hemodiálisis renal.
- Lesión hepatoceliular aguda con ictericia, incluso insuficiencia hepática fulminante.
- Pancreatitis aguda.
- Trombocitopenia, hipotiroidismo.
- El más teratógeno.
- Parkinsonismo.
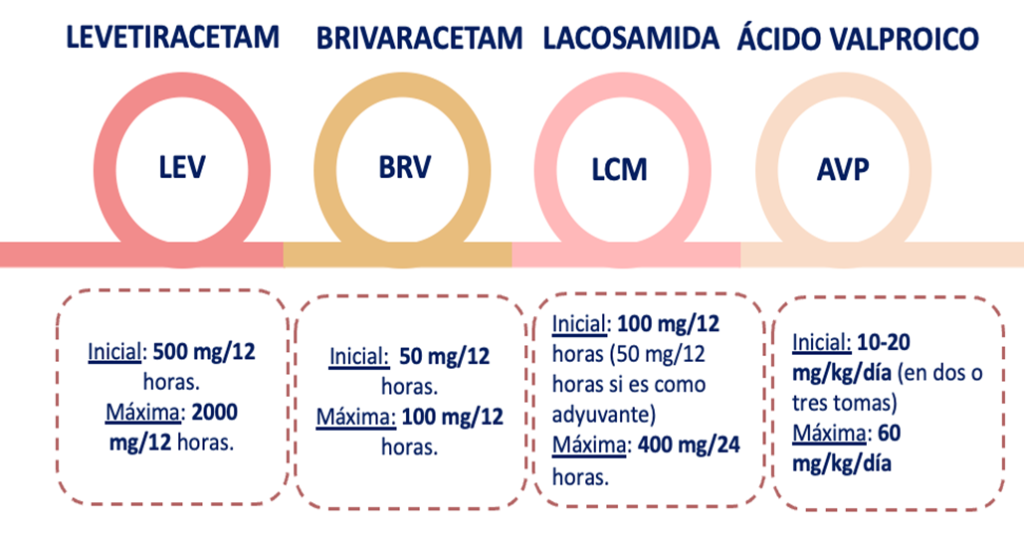
Figura 4. Resumen dosificación de los principales antiepilépticos empleados en anestesiología.
CONCLUSIONES
Durante el perioperatorio existen una gran variedad factores de riesgo para desencadenar un episodio convulsivo. Ante una convulsión es de vital importante realizar un diagnóstico etiológico y corregir la causa. Los antiepilépticos deben mantenerse en el perioperatorio, reiniciándose en el postoperatorio inmediato. Si no es posible la vía oral debemos buscar una vía alternativa (intravenosa, sonda nasogástrica y otros). Es importante que como anestesiólogos conozcamos los efectos adversos y las interacciones de los principales fármacos.
BIBLIOGRAFÍA
- VanHaerents S, Gerard EE. Epilepsy Emergencies: Status Epilepticus, Acute Repetitive Seizures, and Autoimmune Encephalitis. Continuum (Minneap Minn). 2019 Apr;25(2):454-476. Disponible en: doi: 10.1212/CON.0000000000000716. (PubMed)
- Bassel W. Abou-Khalil. Update on Antiseizure Medications 2022. CONTINUUM (MINNEAP MINN). 2022;28 (2, EPILEPSY):500–535. Disponible en: doi: 10.1212/CON.0000000000001104 (PubMed)
- Marciani MG, Gotman J, Andermann F, Olivier A. Patterns of seizure activation after withdrawal of antiepileptic medication. Neurology 1985;35(11): 1537-1543. Disponible en: doi:10.1212/wnl.35.11.1537 (PubMed)
- López-González FJ, Villanueva V, Falip M, Toledo M, Campos D, Serratosa J. Manual de Práctica Clínica en Epilepsia. Madrid: SEN ediciones; 2019. Dispobible en: http://epilepsia.sen.es/wp-content/uploads/2020/06/Recomendaciones-Epilepsia-SEN-2019.pdf
- Manole AM, Sirbu CA, Mititelu MR, Vasiliu O, Lorusso L, Sirbu OM, Ionita Radu F. State of the Art and Challenges in Epilepsy-A Narrative Review. J Pers Med. 2023 Apr 1;13(4):623. Disponible: doi: 10.3390/jpm13040623. (PubMed)
- Nevitt SJ, Sudell M, Cividini S, Marson AG, Tudur Smith C. Antiepileptic drug monotherapy for epilepsy: a network meta-analysis of individual participant data. Cochrane Database Syst Rev. 2022 Apr 1;4(4):CD011412. Disponible: doi: 10.1002/14651858.CD011412.pub4. (PubMed)
- Ghatan S, McGoldrick P, Palmese C, La Vega-Talbott M, Kang H, Kokoszka MA, Goodman RR, Wolf SM. Surgical management of medically refractory epilepsy due to early childhood stroke. J Neurosurg Pediatr. 2014 Jul;14(1):58-67. Disponible en: doi: 10.3171/2014.3.PEDS13440. (PubMed)