 Patricia Ros Sánchez
Patricia Ros Sánchez
Antonio Maldonado Contreras

Cómo citar este artículo: Ros Sánchez, P., & Maldonado Contreras, A. (2012). Caso: Manejo anestésico de pacientes con enfermedad de Motoneurona. Revista Electrónica AnestesiaR, 4(3), 2. https://doi.org/10.30445/rear.v4i3.362
Caso
Mujer de 77 años con AP de HTA, DM tipo II, Insuficiencia renal leve y dislipemia en tratamiento domiciliario con Adiro 100mg/24h, Metformina 850mg/12h, Candesartán 16/12.5mg/24h, Provastatina 20mg/24h, Omeprazol 40mg/24h, Furosemida 40mg/24h y Acido Ursodesoxicólico 300mg/24h. No alergias medicamentosas conocidas.
Se programa en quirófano de Cirugía General para realización de TIROIDECTOMIA TOTAL por diagnóstico de BOCIO COLOIDE GRADO IV CON COMPRESION TRAQUEAL Y ESOFAGICA.
La paciente en Enero del 2.011 comienza a ser estudiada en el Servicio de Neurología por cuadro de disartria de 6 meses de evolución que se acompaña de disfagia y tos frecuente al comer o beber agua.
Se ingresa para estudio, y a la exploración se mantiene consciente y orientada sin afectación de funciones superiores. Movimientos oculares intrínsecos y extrínsecos normales. Campimetría normal. Moderada disartria y disfagia con reflejo nauseoso conservado. Amiotrofia lingual. Sin paresias ni amiotrofias en miembros. ROT conservados. RCP flexores. Sensibilidad algésica conservada y vibratoria aparentemente disminuída en MMII. No dismetría ni dificultad en la marcha.
Se realizan las siguientes pruebas complementarias:
– ANALITICA GENERAL: Glucosa: 95mg/dl (64-107); Urea: 80mg/dl (10-40), Cr: 1´3mg/dl (0´5-1´3); Calcio: 10´6mg/dl (8´5-10´5); PCR: 1.1 (<0´8); Resto del ionograma y función renal normal. HbA1c: 6´8% (<5´7%); Triglicéridos: 184mg/dl (40-170); HDL-colesterol: 36mg/dll (>35); LDL-Colesterol: 130mg/dl; TSH: 0´13 (0´2-5mU/L); T4:1´91ng/dl (0´8-1´9); serie roja, plaquetas, hemoglobina y hematocrito dentro de los parámetros de la normalidad.
– Ac. Antirreceptor Ach negativos.
– TAC CUELLO: Glándula tiroidea muy aumentada de tamaño, de aspecto multinodular, densidad interna heterogénea y contornos lobulados. Cranealmente se extiende entre trapecio derecho y pared faríngea homolateral hasta C2 haciendo prominencia en oro/hipofaringe. En porción media rodea prácticamente por completo estructuras laríngeas y tráquea superior con importante compresión del esófago por parte de ambos lóbulos tiroideos. Caudalmente la glándula llega a mediastino anterosuperior, inmediatamente por encima de la vena innominada.
– RM Craneal: pequeñas lesiones inespecíficas hiperintensas que sugieren origen vascular. Leve dilatación de sistema ventricular y subaracnoideo sugestivo de atrofia. Resto sin interés.
– EMG: pendiente.
Ante la clínica de la paciente y los resultados de las pruebas complementarias es valorada por Cirugía general y Endocrinología: BOCIO GRADO IV CON CLINICA COMPRESIVA. Perfil hormonal tiroideo en consonancia con la gran masa de tejido tiroideo. Dado el gran volumen tiroideo y la clínica compresiva, a pesar de la edad y el aumento de riesgo quirúrgico se decide programar de forma preferente para realización de TIROIDECTOMIA TOTAL, encontrándose los parámetros analíticos y de coagulación dentro de la normalidad en el estudio preanestésico y suspendiendo AAS 7 días antes de la intervención.
Manejo Anestésico Intraoperatorio
Se programa para realización de intubación orotraqueal con fibroscopio óptico flexible con la paciente despierta, administrando sedación mínima para evitar depresión respiratoria dada la clínica de la paciente. Se emplea pulverización de la región nasotraqueal con Lidocaína al 4% y perfusión de Remifentanilo al 5% a una concentración de 100µg/ml y a una velocidad de infusión de 0´05µg/kg/mto. Se intuba sin complicaciones empleándose TOT flexo-metálico nº 7, y a continuación se realiza inducción anestésica con 160mg de Dipriván al 1%, 0.20mg de Fentanilo, 0´7mg de Atropina y 40mg de Rocuronio.
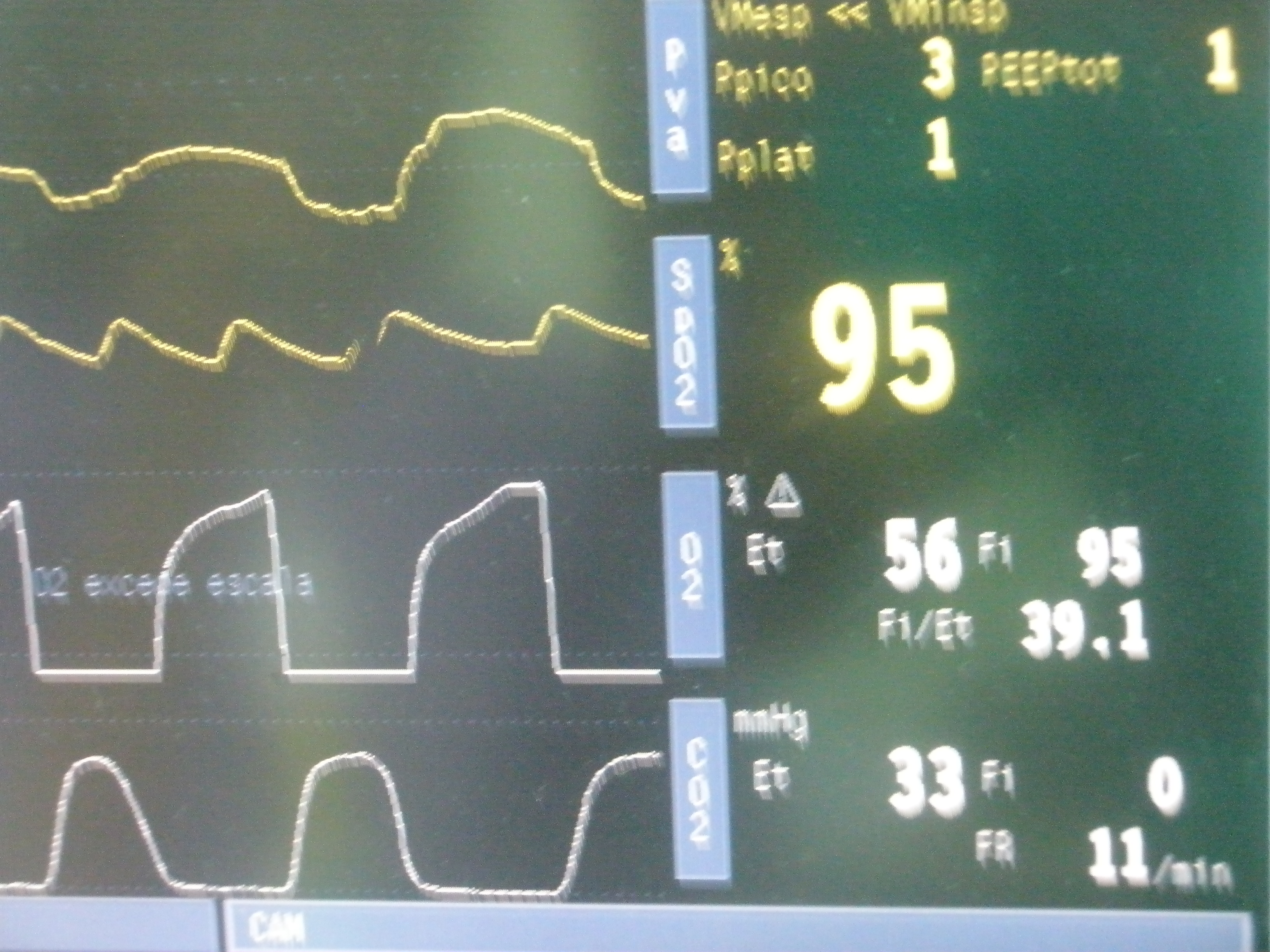
Para el mantenimiento anestésico de la paciente durante la intervención, se emplea Sevofluorano al 1% junto con N2O al 50% + aire, manteniendo de este modo una CAM de 1 y un BIS con valores comprendidos entre 40 y 55. Para analgesia intraoperatoria se administra perfusión de Remifentanilo a dosis comprendidas entre 0´05-0´15 µg/kg/minuto, manteniéndose de este modo TA en torno a 140/70mmHg y FC estable alrededor de los 80lpm. Ventilación controlada modo BIPAP (VT: 500ml; FiO2: 50%; Pr en vías aéreas:20; PEEP:5; FR:14). Se administra para protección gástrica un antiemético (Ondansetrón 4mg) y un antiH2 (Ranitidina 50mg). Para analgesia postoperatoria se comienza a administrar Paracetamol 1g + Dexketoprofeno 50mg. La intervención transcurre sin incidencias intraoperatorias, trasladándose a la paciente a la finalización de la misma a la Sala de Reanimación intubada y sedoanalgesiada para valoración de posible sangrado y compresión traqueal en las 12 horas siguientes.
Es conectada al respirador en modo BIPAP con Psop en 20 que puede ir reduciéndose gradualmente hasta pasar a CEPAP con presión de 10 y PEEP de 5, consiguiendo volúmenes Tidal de 6,7-7l y manteniendo saturaciones de O2 en torno a 96%. Finalmente se pasa a tubo en T (Humivent) con ventilación espontánea, permaneciendo eupneica, con buena dinámica ventilatoria, hemodinámicamente estable sin apoyo vasoactivo, forzando diuresis con Furosemida. No se aprecia sangrado por herida laparotómica ni edematización de región cervical. Por este motivo y dada la buena adaptación de la paciente, se procede a realizar extubación a las 12 horas desde su ingreso, reduciéndose la FiO2 en Ventimask de forma progresiva que tolera con las mismas saturaciones basales con las que llegó al quirófano (90-92%), realizándose alta a sala de Cirugía. Una vez allí sufre cuadro de insuficiencia respiratoria severa que precisa nueva reintubación y VM. Tras la resolución del cuadro se realiza nuevo intento de extubación en las 24 horas siguientes, exitoso aparentemente en las primeras horas, llamando la atención a la exploración neurológica la presencia de disartría, debilidad muscular, fasciculaciones, atrofia generalizada y dificultad para la protusión lingual. Éstos signos junto con el cuadro de insuficiencia respiratoria mantenida en ausencia del efecto de los relajantes musculares, hace tomar la decisión de realizar interconsulta a Neurología quien establece realizar diagnóstico diferencial entre enfermedad de motoneurona bulbar y otros cuadros de debut subagudo vs. agudo, tales como miopatía inflamatoria, miopatía hipotiroidea o rabdomiolisis que se descartan con las pruebas de laboratorio pertinentes y tras completar estudio con EMG (del que estaba pendiente) de forma preferente, que confirma datos de ENFERMEDAD DE MOTONEURA DE PREDOMINIO BULBAR (ELA). A los dos días desde su diagnóstico, dado el cuadro de insuficiencia respiratoria no resuelta, se decide reintubar a la paciente con posterior realización de traqueotomia con conexión a VM y gastrostomía, trasladándose tras hablar con UCI a Unidad de Ventilación Mecánica Domiciliaria.
Conclusión
Se presenta un caso de una paciente con ESCLEROSIS LATERAL AMIOTROFICA (ELA), por la importancia que conlleva un diagnóstico preoperatorio preciso de posibles enfermedades de etiología neuromuscular para evitar posibles complicaciones relacionadas con la debilidad muscular, fundamentalmente de tipo respiratorio y sobre todo en relación con el uso de relajantes musculares. Igualmente se destaca la importancia de realizar un diagnóstico diferencial adecuado, sobre todo con otras enfermedades neuromusculares tipo miastenia gravis o miopatía tiroidea, ya que inicialmente, en esta paciente, síntomas tales como la disfagia y dificultad respiratoria se atribuían a la compresión esofágica y traqueal por bocio coloide gigante, pudiendo hacerse uso inadecuado de los fármacos anestésicos así como una incorrecta extubación si no se sospecha y se conoce la etiología y evolución de las diferentes enfermedades neuromusculares.
Revisión del manejo anestésico en patología neuromuscular
Los trastornos neuromusculares, aunque raramente se encuentran en la práctica anestésica habitual, constituyen un grupo de patologías que desafían el tratamiento perioperatorio y de cuidados intensivos. La aplicación de protocolos de manejo facilitará la toma de decisiones eficaces y ajustadas a la evolución del paciente. Los trastornos neuromusculares tienen un potencial significativo de interactuar con un plan anestésico inadecuado, y todos los pacientes requieren una atención perioperatoria especial respecto al tratamiento anestésico. Las situaciones de catástrofe en enfermedades neuromusculares, son aquellas en las que su curso clínico es susceptible de causar riesgo de muerte para el paciente, pudiendo este desenlace estar relacionado con tres supuestos:
1.- Insuficiencia respiratoria aguda de origen neuromuscular.
2.- Disautonomía.
3.- Anestesia general o regional.
Las lesiones musculares en muchas ocasiones se encuentran ligadas a una lesión concomitante de tipo respiratorio o cardiovascular, que junto a la disminución de la fuerza muscular y una mayor sensibilidad a ciertos fármacos anestésicos pueden predisponer al fallo respiratorio y complicaciones potencialmente graves en el periodo perioperatorio.
Para el paciente miópata en general, la utilización indiscriminada de ciertos fármacos, en especial relajantes musculares, puede ser responsable de una respuesta habitualmente exagerada que se caracteriza por parálisis prolongada o hiperpotasemia, insuficiencia respiratoria, rigidez muscular, e incluso en pacientes susceptibles hipertermia maligna.
Sin duda, la identificación y valoración preoperatoria de estos pacientes así como la identificación de los grupos musculares afectados y su magnitud, es de vital importancia, debiendo realizar una historia clínica detallada con el informe neurológico documentado así como con las correspondientes pruebas complementarias.
Trastornos de Motoneurona
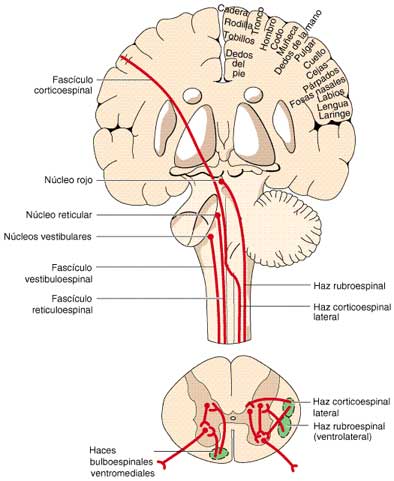
Afectan a las motoneuronas superiores o inferiores de la corteza cerebral, tronco del encéfalo y médula espinal. La Esclerosis Lateral Amiotrófica (ELA), es la enfermedad más frecuente dentro de este grupo y afecta a las motoneuronas superiores e inferiores. Otros ejemplos de enfermedad de motoneurona son la atrofia muscular espinobulbar (Enfermedad de Kennedy), Ataxia de Friedreich (mixta de motoneuronas superiores e inferiores) y la Atrofia Muscular espinal (motoneuronas inferiores).
La ELA (Enfermedad de Lou Gehrig) se caracteriza por una pérdida progresiva de motoneuronas dentro de la corteza cerebral, los núcleos bulbares de los nervios craneales y los núcleos del cuerno ventral de la médula espinal, lo que conduce a debilidad muscular progresiva, atrofia muscular y pérdida de la masa neuronal en estas localizaciones. Las funciones sensitivas, incluidas la capacidad intelectual y la función cognitiva, así como la función vesical e intestinal, no suelen verse afectadas.
Tiene una incidencia de 2 por cada 100.000 habitantes y suele comenzar entre los 40 y los 50 años, con más varones afectados que mujeres. La mayoría de los casos son esporádicos y existen formas familiares raras (autosómica dominante y recesiva). La etiología de esta enfermedad no está clara, pero parece ser que mutaciones en la superóxido dismutasa dan lugar a una mayor formación de radicales libres. El diagnóstico se realiza mediante electromiografía y electroneurografía, así como mediante la exploración neurológica que muestra debilidad espástica temprana de las extremidades superiores e inferiores, fasciculaciones musculares subcutáneas típicas y afectación bulbar que influye en la función faríngea, el habla y la musculatura facial.
Consideraciones Anestésicas
La afectación bulbar combinada con debilidad de la musculatura respiratoria conduce a un riesgo de aspiración y complicaciones pulmonares. Estos pacientes pueden tener una mayor sensibilidad a los efectos depresivos respiratorios de los sedantes e hipnóticos, existiendo informes de hiperreactividad simpática y fallo autónomo. Debe evitarse el SUXAMETONIO debido a riesgo de hiperpotasemia provocado por la denervación e inmovilización. Los RELAJANTES MUSCULARES NO DESPOLARIZANTES pueden causar un bloqueo muscular prolongado o pronunciado debiendo usarse con mucha precaución. Se ha usado la anestesia general combinada con la epidural sin complicaciones.
Patricia Ros Sánchez
Unidad de Anestesiología y Reanimación del Hospital Clínico“ San Cecilio“.
Antonio Maldonado Contreras
Unidad de Gestión Clínica de Enfermedades neurológicas del Complejo Hospitalario de Jaén.

a mi papá le diagnosticaron una enfermedad motoneuronal {ela} queria saber si hay algun tratamiento ya que aqui en URUGUAY no hay nada para hacerle-agradezco contestacion por favor
Aún no existe (en ninguna parte del mundo) un tratamiento para ELA que se haya comprobado mediante estudios que mejore los sintomas o prolongue la vida en forma significativa. Riluzole es la única medicina que se está usando pero no mejora los sintomas y solo prolonga la vida un promedio de 3 o 4 meses. Cuando a alguien le diagnostican ELA, siempre es bueno tener una segunda o tercera opinión ya que en algunos casos puede ser otra enfermedad tratable que fácilmente confundida con ELA. Por lo general se hace lo que se conoce como «diagnóstico diferencial» lo que significa descartar cualquier otra enfermedad que se parezca a ELA.